IRB Study Reporting
In response to the regulatory obligation, the HU IRB, in conjunction with the Institutional Biosafety Committee (IBC), utilizes a three-category reporting system to facilitate review of reports and determinations about whether a reported problem/event raises new concerns about risk to participants or others; the risk/benefit ratio; the approved informed consent document; and the need for re-consent.
-
Prompt Reporting is required for unanticipated problems involving risk to participants or others (including unanticipated serious or life-threatening adverse events) and anticipated or unanticipated deaths. Prompt reports should be submitted to the IRB within 48 hours of when the PI is made aware of the the event.
-
Non-Prompt Reporting is required for anticipated problems/anticipated serious adverse events or unrelated deaths (required by sponsor but not by HU). Non-Prompt reports should be submitted to the IRB within 7-14 days of when the PI is made aware of the the event.
-
Continuation Review Reporting is required if any problems/adverse events occurred within 12 months prior to the continuation review request. A written summary of all problems/adverse events involving subjects since the study was initiated, whether anticipated or unanticipated, serious or not serious, life-threatening or not life-threatening, or related or not related should be included in the study continuation request.
Adverse Events
All unanticipated problems and adverse events must be reported to the IRB by submitting a SAE Reporting Form in the iRIS platform.
An internal event/problem is one that occurs with research subjects enrolled in a project approved by the HU IRB and directed by an investigator employed by the University or one whose project is under the purview of the HU IRB.
An external event/problem is one that occurs with research subjects enrolled in multi-center research projects that do not fall under the purview of the HU IRB.
Institutional policy requires investigators to report deaths that are related to the study procedures immediately upon the investigator’s receipt of the information (i.e., within 48 hours). The PI includes reports of deaths that are not related to the study procedures (i.e., due to underlying disease progression) in the summary of problems/adverse events submitted at the time of IRB continuation review
The PI reports unanticipated life-threatening experiences within 7 calendar days of his/her receipt of the information and all other serious and unanticipated events/problems within 14 calendar days of his/her receipt of the information. Institutional policy requires the investigator to provide follow-up reports on serious or life-threatening and unanticipated and related events within 14 calendar days of his/her receipt of the information.
Protocol Violation
A protocol violation is any exception or deviation involving a single participant that is not approved by the IRB prior to its initiation or implementation. These protocol violations may be major or minor violations. All protocol violations must be reported to the IRB by submitting a Notice of Protocol Development/Violation form in the iRIS platform.
If the investigator makes study changes (i.e., modifications, exceptions or deviations) to eliminate apparent hazards to the participant(s) without prior IRB approval, the investigator must immediately report the changes to the IRB for review and a determination as to whether the changes are consistent with the participant’s continued welfare.
Exceptions or deviations are changes that impact individual participants and do not change the overall study. Investigators may not initiate these changes without prior IRB review and approval, except where necessary to eliminate apparent hazards to the participant.
-
The IRB considers enrollment of a research participant in a study that fails to meet current IRB approved study inclusion criteria or falls under study exclusion criteria to be a study exception.
-
The IRB considers a departure from the current IRB approved procedures that impact an individual participant to be a protocol deviation.
Quick Links
Major Violations
A major violation is one that may impact participant safety, make a substantial alteration to risks to participants, or any factor determined by an IRB member as warranting review of the violation by the convened IRB. Examples of major violations may include, but are not limited to:
-
Failure to obtain informed consent, i.e., there is no documentation of informed consent, or informed consent is obtained after initiation of study procedures;
-
Enrollment of a participant who did not meet all inclusion/exclusion criteria;
-
Performing study procedures not approved by the IRB;
-
Failure to report serious unanticipated problems/adverse events involving risks to participants to the IRB and (if applicable), the sponsor;
-
Failure to perform a required lab test that, in the opinion of the PI, may affect participant safety or data integrity;
-
Drug/study medication dispensing or dosing error;
-
Study visit conducted outside of required time frame that, in the opinion of the PI or IRB, may affect participant safety;
-
Failure to follow safety monitoring plan.
-
Use of invalid consent form, i.e., consent form without IRB approval stamp or outdated/expired consent form;
-
Enrollment of participants after IRB-approval of study expired or lapsed;
Minor Violations
A minor violation is a violation that does not impact participant safety or does not substantially alter risks to participants. Examples of minor violations may include, but are not limited to:
-
Implementation of unapproved recruitment procedures;
-
Missing original signed and dated consent form (only a photocopy available);
-
Missing pages of executed consent form;
-
Inappropriate documentation of informed consent, including:
-
Missing participant signature;
-
Missing investigator signature;
-
Copy not given to the person signing the form;
-
Someone other than the participant dated the consent form;
-
Individuals obtaining informed consent not listed on IRB approved study personnel list.
-
-
Over-enrollment;
-
Failure to submit continuing review application to the IRB before study expiration.
Approved Study Procedure Violations
-
Failure to follow the approved study procedure that, in the opinion of the PI, does not affect participant safety or data integrity;
-
Study procedure conducted out of sequence;
-
Omitting an approved portion of the protocol;
-
Failure to perform a required lab test;
-
Missing lab results;
-
Enrollment of ineligible participant (e.g., participant’s age was 6 months above age limit);
-
Study visit conducted outside of required timeframe;
-
Submission Checklist
A complete submission increases the chance of approval. The following information should be included when submitting an adverse event or study protocol violation report:
-
A complete Notification of Protocol Development/Violation Form or SAE (Serious Adverse Event) Reporting Form. Responses should be clear and concise to improve readability and understanding. Complete all fields. Fields that are not applicable to the research should indicate this when possible.
-
Supplemental Documentation. Documentation that supplements or verifies information included in the Notification of Protocol Development/Violation Form or SAE (Serious Adverse Event) Reporting Form.
-
Sponsor Documentation. If applicable, provide correspondence from the study sponsor related to the adverse event or protocol violation.
Education and Resources
Education
Resources
Forms and Tools
- iRIS Platform
- Ethical IRB Guidelines and Principles
- GCP Training Requirement
- Guidance for Case Reports
- Guidance for Faculty and Research Staff
- Guidance on Recruitment Material
- Guidance: Student Research Guide
- IRB Charge Policy
- IRB SOP
- IRB SOP Supplement
- IRB SOP DoD Addendum
- Limited IRB Review
- Review of PHI-Related Protocols
- Template: Assent Form (Download)
- Template: Informed Consent Form (Download)
- Template: Interview Preamble (Download)
- Template: Survey Preamble (Download)
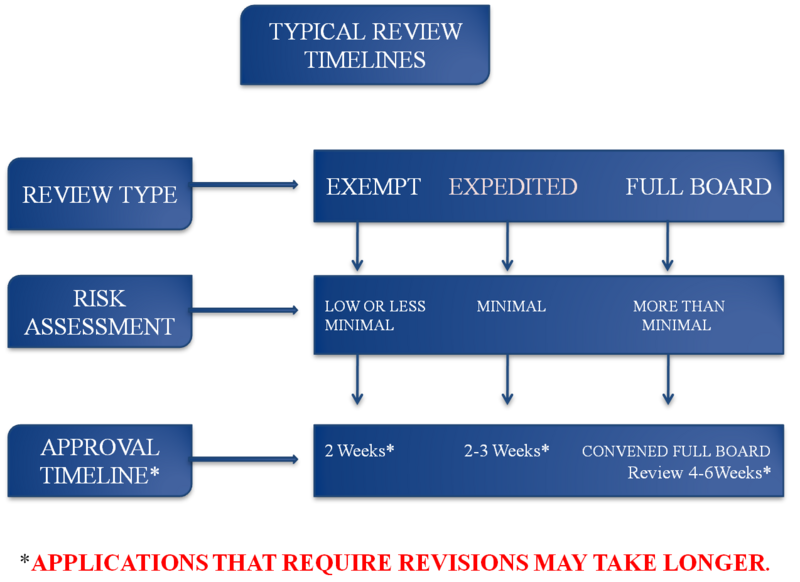
- Typical IRB Review Timelines
{kind=link}
Links
- 45 CFR 46: The Common Rule | eCFR
- 21 CFR 50: FDA regulations on the protection of human subjects | eCFR
- 21 CFR 56: FDA regulations on Institutional Review Boards (IRBs) | eCFR
- A How to Guide: The Use of Social Media in Research | NIH
- Belmont Report
- Human Research Protection Program Resources | HHS
- Office for Human Research Protections (OHRP) | HHS
ORRC staff serves Howard University faculty, staff, and students by helping them ensure that their research and teaching modules comply with all applicable federal, state, and local regulations and policies, as well as Howard University policies. ORRC staff are available to answer individual inquiries, meet with researchers and instructors, and to present and facilitate educational workshops and trainings.