Getting Started with the IRB
Prior to submitting an application to the IRB, it is necessary to know if the project requires IRB approval, how to prepare to submit an application to the IRB, and when an IRB application should be submitted.
What Activities Require IRB Approval?
Guided by the OHRP Guidance at 45 CFR 46.102(d); and the FDA Guidance at (21 CFR 50.3(c), 21 CFR 56.103(c), 21 CFR 312.3(b), and 21 CFR 812.3(h)), IRB Approval is required for projects that are defined as "Research" or a "Clinical Investigation" and involves human participants/subjects.
Is The Project Research?
IRB review is required for project activities that are considered “Research” as defined by the OHRP guidance at 45 CFR 46.102(d) and other federal agencies. Based on this definition, Research is a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge.
Activities that meet this definition constitute research for purposes of federal policy. This qualification is regardless of whether project activity is conducted or supported under a program that is considered research for other purposes. For example, some demonstration and service programs may include research activities.
For clarification, the following definitions are provided:
- Systematic Investigation: An activity that plans (prospectively) to incorporate data collection (quantitative or qualitative) and data analysis to answer a question. Activities are not research if they do not involve a systematic approach involving a predetermined method for studying a specific topic, answering a specific question, testing a specific hypothesis, or developing theory.
- Generalizable Knowledge: Activities designed (with intent) to develop or contribute to generalizable knowledge are those designed to draw general conclusions, inform policy, or generalize findings beyond a single individual or an internal program (e.g., publication or presentation). The intent to develop or contribute to generalizable knowledge makes an activity research. Results do not have to be published or presented, to qualify the activity as research.
Examples of activities that are typically considered systematic investigations include interviews, focus groups, surveys, questionnaires, analysis of data and specimens, observational studies, epidemiological studies, or the review of medical records as part of systematic investigation.
For more information on research determination, see the OHRP Guidance at 45 CFR 46.102(d).
Is The Project A Clinical Investigation?
Clinical investigations are considered Research and require IRB review and approval. FDA guidance defines a clinical investigation as any experiment that:
- Involves a test article and one or more human participants/subjects, and that
- Either is subject to the requirements for prior submission to the FDA under section 505(i) or 520(g) of the FD&C Act, or
- Need not be subject to the requirements for prior submission to the FDA under relevant sections of the Act, but the results of which are intended to be later submitted or held for inspection by the FDA as part of an application for a research or marketing permit (21 CFR 50.3(c), 21 CFR 56.103(c), 21 CFR 312.3(b), and 21 CFR 812.3(h)).
For clarification, a test Article is any drug, biological product, medical device, human food additive, color additive, electronic product, or any other article for human use that is subject to FDA regulations.
Examples of activities that are clinical investigations include clinical trials that involve investigational drugs or devices, research testing the safety and effectiveness of a device, or medical outcome studies comparing approved drugs or devices.
For more information on clinical trial investigation determination, see the FDA Guidance at (21 CFR 50.3(c), 21 CFR 56.103(c), 21 CFR 312.3(b), and 21 CFR 812.3(h)).
Does The Research Involve "Human Subjects/Participants"?
IRB approval is required for research or clinical trials that involve human participants/subjects. Once the study has been determined as “Research”, the next step is to determine if human subjects/participants are involved.
A human subject/participant is a living individual about whom research is conducted through:
- The collection of information or biospecimens through intervention or interaction with the individual for use, study, or analysis, or
- The acquisition, use, study, analysis, or generation of identifiable private information or identifiable biological material.
The OHRP Chart 01: Is an Activity Human Subjects Research Covered by 45 CFR Part 46? is available to assist with determining if a research project involves human subjects/participants.
Exceptions and Exclusions
Projects that are not defined as “Research” are excluded from federal regulation 45 CFR 46 “Protection of Human Subjects,” also known as the Common Rule and, thus, are excluded from the requirements of IRB review and approval. However, the Howard University Graduate School and some project sponsors may request an official letter from the IRB that verifies this determination. In these cases, it is required that an IRB application is submitted for verification.
Studies in basic and applied sciences such as Engineering, Physics, Computer Sciences, Chemistry, and others that are defined as “Research,” but do not involve interactions or interventions with human participants, biohazards or animals are usually excluded from the requirements for IRB review and may qualify for an Exclusion review. Regardless of discipline, all exclusion reviews requested by student applicants must include a RCR Workshop certification of completion from the Graduate School.
Studies that do not fall under the definition of "Research" and do not involve human participants are considered Exceptions of the requirements for IRB review and do not require a submission to the IRB. However, it is recommended to contact the ORRC to validate this. Studies that are Exceptions from IRB review requirements include:
- Oral histories and journalistic activities designed exclusively to create a record of specific individuals or events.
- Training activities when they are NOT intended to contribute to generalizable knowledge.
- Classroom activities where the objective of the activity is to teach proficiency in performing certain tasks or using specific tools or methods, when the activity is NOT intended to contribute to generalizable knowledge.
- Biographies.
- Service or course evaluations.
- Services, courses, or concepts where the results are NOT intended to be shared beyond the Howard University Community.
- Classroom exercises specifically designed to fulfill course requirements or to train students in the use of specific methods or devices.
- Quality assurance activities designed to continuously improve the quality or performance of a department or program, and there is NO intention to share the results beyond the Howard University Community.
- Case reports, which are a retrospective analysis of one, two, or three clinical cases that describe an interesting treatment, presentation, or outcome. More on case studies and case reports can be found at Case Report Guidance.
Training Requirements
All personnel listed in a research study, regardless of their position, must complete the web-based training through the Collaborative Institutional Training Initiative (CITI) Program. Visit the CITI Program page for more information on training requirements.
CITI ProgramWhich IRB To Use?
Howard University has two IRBs. One that reviews studies with a biomedical focus, and one that reviews all other studies. Each IRB is explained below.
Medical IRB
The Medical IRB is generally charged with reviewing research applications that originate in the Health Sciences division along with research study protocols that involve medical procedures, devices, and drugs. All research records and staff are accountable to external review and monitoring by funding agencies.
The Medical IRB meets every other Wednesday. Schedule meeting dates and submission deadline dates may be found here.
Non-Medical IRB
The Non-Medical IRB typically reviews those research applications that originate from the Academic Affairs division along with research studies that are generally socio-behavioral in scope.
The Non-Medical IRB meets every other Wednesday. Schedule meeting dates and submission deadline dates may be found here.
Central and Single IRBs
HU IRBs may cede the ethical oversight of a research study to a central or single IRB. Investigators must confirm with the ORRC before submitting any protocol to a central or single IRB.
- A central IRB is specifically established to conduct the ethical review for all sites participating in a program that usually includes more than one multi-site study. (e.g. NCI Central IRB)
- A single IRB (sIRB) is the selected IRB of record that conducts the ethical review for one or more sites participating in cooperative research. (e.g. GHUCCTS IRB)
More information regarding central or single IRBs can be found here.
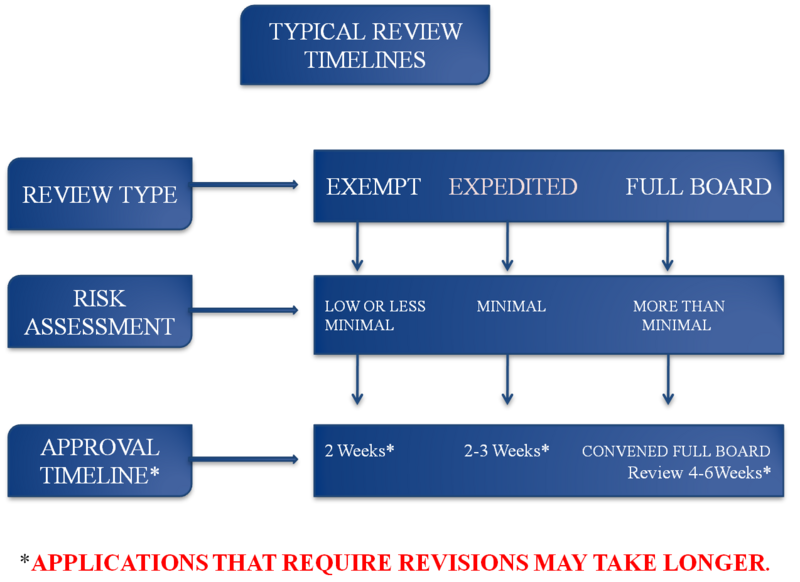
IRB Review Types
Qualifications for Exempt Review
Research that qualifies for exempt status is considered to pose minimal risk to participants and falls into a specific category defined by federal regulation 45 CFR 46.104.
Exempt Reviews are not subject to the same regulatory requirements as Expedited or Full Board reviews. They are reviewed outside of the scheduled IRB meetings, and can be submitted at any time of the year.
The IRB has the authority to make a final determination as to whether a study meets the qualifications for Exempt Review and can recommend that a study is resubmitted as for Expedited or Full Board Review. At least one designated IRB member will review the study submission to determine if it qualifies for Exempt review and that all compliance standards for Minimal Risk are met. The timeline for this type of review is generally up to 2 weeks. Study submissions may be tracked in iRIS, the electronic compliance platform. The ORRC may be contacted if further information is necessary.
Qualifications for Expedited Review
For a study to qualify for Expedited review, it must meet the definition of “Minimal Risk” as defined by 45 CFR 46, it must be excluded from any of the categories for exemption, and it must fall into one of the Expedited Review Categories.
Expedited Review does not mean “faster review”. Studies that qualify for Expedited Review are not subject to the same regulatory requirements as Full Board Reviews. Like Exempt Reviews, Expedited Reviews are reviewed outside of the scheduled IRB meetings, and can be submitted at any time.
The IRB has the authority to make a final determination as to whether a study meets the qualifications for Expedited Review and can recommend that a study be resubmitted for Exempt or Full Board Review. At least one designated IRB member will review the study submission to determine if the study qualifies for Expedited Review and that all compliance standards for Minimal Risk are met. The timeline for this type of review is generally up to 3 weeks. Study submissions may be tracked in iRIS, the electronic compliance platform. The ORRC may be contacted if further information is necessary.
Qualifications for Full Board Review
Studies that are greater than minimal risk or minimal risk studies that include vulnerable populations or investigational procedures or devices are reviewed through the Full Board review process. If a study submission does not qualify for Exempt or Expedited review, it will be reviewed by the Full Board.
The IRB has the authority to make a final determination as to whether a study meets the qualifications for Full Board Review and can recommend that a study be resubmitted for an Exempt or Expedited Review. Full Board Reviews are reviewed at convened IRB Meetings. Meeting schedules for the Medical and Non-Medical IRB are provided on the Human Research page of this website. The timeline for this type of review is generally up to 6 weeks. Study submissions may be tracked in iRIS, the electronic compliance platform. The ORRC may be contacted if further information is necessary.
IRB Submission Dates for Graduate Students
The IRBs will review submissions received throughout the year. However, specific deadlines apply for IRB submissions that are needed for candidacy or any other Graduate School requirements.
Fall Term
Submit the final version of the IRB Application by: the last Monday in August
Submit Candidacy Applications by: the last Monday in November
Spring Term
Submit the final version of the IRB Application by: the last Monday in January
Submit Candidacy Applications by: the last Monday in April
Summer Term
Submit the final version of the IRB Application by: the 2nd Monday in April
Submit Candidacy Applications by: the 2nd Monday in July
Informed Consent and Assent
Informed consent is a cornerstone of ethical research and a legal requirement under federal regulations (21 CFR 50; 45 CFR 46). It is not just a signed form, but an ongoing educational process between the investigator and the participant. This process ensures that individuals understand:
- The purpose, procedures, risks, and benefits of the research.
- That participation is voluntary and can be withdrawn at any time without penalty.
- How their privacy and rights will be protected.
The consent process must use language understandable to the participant and minimize coercion or undue influence. It should provide sufficient detail for a reasonable person to make an informed decision and must never include language that waives legal rights or releases liability.
Assent is equally important when involving children or individuals with impaired decision-making capacity. Assent is an affirmative agreement to participate in research. Federal regulations require both assent from the individual and permission from a parent or Legally Authorized Representative (LAR), ensuring respect for autonomy at all developmental levels.
Failure to properly obtain and document informed consent or assent can result in protocol violations, regulatory noncompliance, and harm to participants. Therefore, investigators must treat these processes as essential safeguards for participant rights and welfare, and as a foundation for trust in research.
Preambles in Surveys and Interviews
When research involves surveys, interviews, or questionnaires, investigators should include a preamble at the beginning of the instrument. The preamble serves as a simplified consent statement and should:
- Clearly state that the activity is part of a research study.
- Explain the purpose of the survey/interview and approximate time required.
- Inform participants that their participation is voluntary and they may skip any question or stop at any time.
- Describe how confidentiality will be maintained and whether responses will be anonymous.
- Provide contact information for questions about the study or participant rights.
For minimal-risk studies, a preamble often satisfies the requirement for informed consent when a signed form when a request to waive informed consent is approved to the IRB. However, the language must still meet federal and institutional standards for clarity and transparency.
The ORRC provides informed consent document templates for Investigators to use in their research involving human participants or as guides to create their own documents. The templates contain all required elements of informed consent and additional IRB requirements for HU research involving human participants. Investigators may use these as guides to creating their own consent documents.
The ORRC provides consent preamble templates for interviews and surveys that may be used by Investigators in their research. The templates contain all required elements for informed consent and additional IRB requirements for HU research involving human participants. Investigators may use these as guides to creating their own preambles.
iRIS Access
The iRIS platform (also known as iMedRIS) is used to create and manage studies that require regulatory compliance review. All studies that require IRB approval must be submitted for review in iRIS. Howard University personnel and students who require iRIS access may request an iRIS account by completing the iRIS User Access Request Form. Contact the ORRC for assistance with providing iRIS access to non-HU study personnel. Visit the iRIS page for more information, including virtual iRIS demo/training sessions.
eCompliance - iRISEducation and Resources
Education
Resources
Forms and Tools
- iRIS Platform
- AI Policy
- Ethical IRB Guidelines and Principles
- GCP Training Requirement
- Guidance for Case Reports
- Guidance for Faculty and Research Staff
- Guidance on Recruitment Material
- Guidance: Student Research Guide
- IRB Charge Policy
- IRB Communication with the RSC
- IRB SOP
- IRB SOP Supplement
- IRB SOP DoD Addendum
- Limited IRB Review
- Review of PHI-Related Protocols
- Template: Assent Form (Download)
- Template: Informed Consent Form (Download)
- Template: Interview Preamble (Download)
- Template: Survey Preamble (Download)
- Typical IRB Review Timelines
{kind=link}
Links
- 45 CFR 46: The Common Rule | eCFR
- 21 CFR 50: FDA regulations on the protection of human subjects | eCFR
- 21 CFR 56: FDA regulations on Institutional Review Boards (IRBs) | eCFR
- A How to Guide: The Use of Social Media in Research | NIH
- Belmont Report
- Human Research Protection Program Resources | HHS
- Office for Human Research Protections (OHRP) | HHS
ORRC staff serves Howard University faculty, staff, and students by helping them ensure that their research and teaching modules comply with all applicable federal, state, and local regulations and policies, as well as Howard University policies. ORRC staff are available to answer individual inquiries, meet with researchers and instructors, and to present and facilitate educational workshops and trainings.