Initial IRB Submission
The initial IRB Application is intended to detail the proposed research or teaching activities that involve human participants. The risk level to participants and nature of the research determine the process in which the application will be reviewed.
Risk levels
Minimal Risk or No Greater Than Minimal Risk: According to 45 CFR 46, a study is considered minimal risk when, “the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.”
Greater than Minimal Risk: Studies that do not qualify as "minimal risk" and usually involve medical procedures or devices or create some high degree of discomfort for participants are considered greater than minimal risk studies. This discomfort can be physical, emotional, social, and/or psychological.
Submission Review Process
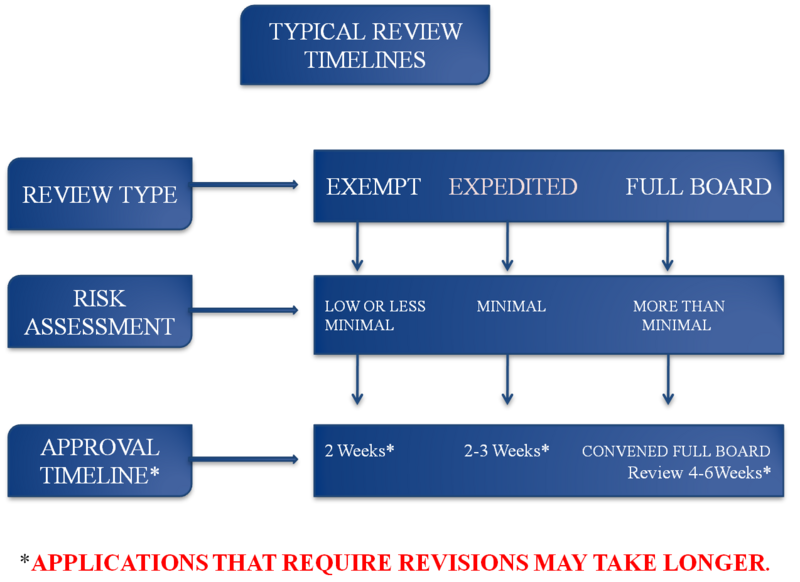
All study submissions to the IRB will undergo one of three review processes (Exempt, Expedited, or Full Board) depending on the nature of the research and the risk to potential participants. Often, the determination of a study submission review process is a judgement call rather than a hard-lined regulatory decision. However, the call is always grounded on the fundamental principle of human subject/ participant research outlined in the Ethical Principles of the Belmont Report.
Respect for Person
-
Treat Individuals as autonomous agents.
-
Protect persons with diminished autonomy. In general, respect for person is an important requirement for IRB approval of research and premised on:
-
Voluntary consent to participate in research
-
Informed consent to participate in research
-
Protection of privacy and confidentiality
-
The right to withdraw from research participation without penalty
-
Beneficence
-
Do unto others as you would have them do unto you.
-
Are the research participants treated the way that you would like to be treated in this situation?
-
Are the risks of research justified by potential benefit to the individual and/or society?
-
Does the study design minimize risk and maximize the potential for benefit?
-
Are conflicts of interest managed so that bias in important judgements related to research conduct are unlikely?
-
Justice
-
Distribute the risk and potential benefits of research equally among those who may benefit from the research.
-
The potential risks of research should be borne equally by the members
-
The research project should not systematically select specific classes or types of individuals simply because of their ease of availability or their compromised positions as opposed to reasons directly related to those of our society who are likely to benefit from the problem being studied.
-
The researcher project does not exclude specific class or type of person who is likely to benefit from research participation or in whom the results of a specific kind of research are likely to be applied.
-
IRB Review Process
Pre-Submission
The PI or a designated member of the study team completes a submission form (for Initial, Study Continuation, Study Modification/Amendment, Study Reporting, or Study Closeout) in the iRIS electronic compliance platform. The form must be reviewed and approved by the IRB before the requested study activity can commence.
ORRC Pre-Review
The ORRC receives the submission and reviews it for completeness and initial adherence to federal, state, and local regulations. If any information is missing from the submission, the PI and any authorized study personnel will be notified and asked to make corrections. If the submission is complete, it will be assigned to a review process and to an IRB member(s) for review. If the submission requires review by the full Committee, the ORRC schedules the submission for review at the next available convened Full Board IRB meeting.
IRB Review
The submission is reviewed by at least one IRB member or in some cases, by a member of the ORRC. This review process can take up to 6 weeks depending on the type of review, complexity of the study, completeness of the submission, and the availability of reviewers.
- Exempt Reviews may take up to 2 weeks
- Expedited Reviews (not to be mistaken with "quick" reviews) may take up to 3 weeks
- Full Board reviews may take up to 6 weeks
Projects that are not "Research" as defined by federal guidance or excluded from the requirements of IRB oversight are not reviewed by the IRB.
Post Review
Once the review of the submission is complete, an outcome letter will be sent to the study PI and authorized study personnel indicating that the submission was approved/accepted, disapproved/unaccepted, requires revisions, may be approved pending the administrative review of given conditions, or the review has been tabled for a given reason. Depending on the outcome, further information may be requested prior to the full approval of the submission.
Data and Safety Monitoring Plan(s) (DSMP)
The IRBs review of Data and Safety Monitoring Plan(s) (DSMP) ensures adequate protection are in place for research participants. Investigators develop DSMP as a mechanism for assuring the safety of human participants and human research data, the validity of data, and the appropriate termination of studies. The IRB requires review and approval of DSMPs for greater than minimal risk research, or clinical investigations funded by the National Institutes of Health (NIH) or regulated by the Food and Drug Administration (FDA).
DSMPs include:
- Plans for monitoring the progress of trials and the safety of subjects;
- Plans for assuring compliance with requirements regarding the reporting of adverse events;
- Plans for review or analysis of cumulative safety data to determine whether harm is occurring;
- Plans for assuring that any action resulting in a temporary or permanent suspension of a clinical trial is reported to the appropriate agencies;
- Plans for assuring data accuracy and protocol compliance;
- Plans for assuring communication among multi-center sites adequately protect the subjects (for multicenter studies where the lead PI is employed by HU or HU is the coordinating institution)
iRIS Access
The iRIS platform (also known as iMedRIS) is used to create and manage studies that require regulatory compliance review. All studies that require IRB approval must be submitted for review in iRIS. Howard University personnel and students who require iRIS access may request an iRIS account by completing the iRIS User Access Request Form. To provide iRIS access to non-HU study personnel, please contact the ORRC. For more information on how to access and use the iRIS platform, visit the iRIS section of this website.
eCompliance - iRISA complete initial IRB submission increases the chance of approval. The initial IRB Application should be accompanied by documents that support the information provided in the application.
The following information should be included with an initial submission for IRB review.
General Requirements
- All research personnel and their study role must be defined in the IRB Application. Please note that Student researchers cannot be listed as the PI. A full-time faculty member must agree to accept responsibility for the project and serve as the PI. The student researcher must then be identified as the Student Investigator (SI) or Co-PI.
- The C/V, biosketch, or resume of ALL study personnel.
- The Department Chairperson (or other department lead) and Dean (or Associate Dean for Research) must approve the initial submission. These individuals must be defined in the IRB Application so that their approval may be requested. If either of these individuals are unable to review the submission, then a designated signatory must be defined in the IRB Application.
- CITI training certificates of completion of ALL study personnel for ALL required CITI modules. For more information on CITI training requirements, visit the CITI page.
- A copy of the project proposal or study protocol that details the study. The information in the IRB Application should coincide with the information in the project proposal or study protocol.
- Letters of support from entities providing resources or services for the study.
- A Consent Form, if adults (age 18 and older) will have to provide their consent to participate in the study. More than one consent form may be used for a study. If necessary, download and use the ORRC Informed Consent Form Template.
- An Assent Form, if children (under 18 years of age) will be consented to participate in the study. A consent form for the parent, guardian, or other Legal Authorized Representative (LAR) of the child is also required. If necessary, download and use the ORRC Assent Form Template.
- A copy of all Recruitment Material. This includes flyers, brochures, emails, etc.
- A copy of all surveys, questionnaires, and other tools that will be used in the study. If necessary, download and use the consent preamble templates for interviews or surveys.
- A copy of all other participant-facing documentation.
- Non-English language documents. If non-English language documents are being used, provide an English-language version of those documents and, for each document, a certificate of translation from English to the necessary language. Entities that provide certified translation services may be found online.
- The status of any ancillary reviews. A letter of approval or statement of the current status of the submission is necessary. Research that involves the care or use of animals requires (IACUC) review and approval. Research that involves biohazardous material requires (IBC) review and approval. Research that involves the use of ionizing radiation requires Radiation Safety Committee review and approval. Research that involves export control concerns requires Export Control Committee review and approval.
Additional Requirements for Student Research
- The Student Investigator and Principal Investigator must be clearly defined in the IRB Application. The role of Principal Investigator should only be assigned to active HU Faculty and certain staff members. Students should not list themselves as Principal Investigators on a study.
- Certificate of completion from the RCR Workshop, if the study is related to a thesis or dissertation. This workshop is a collaborative training requirement presented by the ORRC and the Graduate School. All students seeking candidacy must complete this workshop. Please note that this workshop is separate from the RCR CITI training module. For more information on the RCR workshop, visit the Graduate School website.
- A copy of the student thesis or dissertation, only if the study is related to a student thesis or dissertation.
- A copy of the department approval sheet, only if the study is related to a student thesis or dissertation. This document must be signed by the approval committee and must be included with the thesis or dissertation.
Additional Requirements for Collaborative Research
- A copy of the reliance agreement between HU and the collaborating entity, if HU is collaborating with another entity for the study. For more information visit the reliance(single/central IRB) page.
- A copy of the reliance agreement between HU and the collaborating entity, is ceding the ethical review of the study to another entity. For more information visit the reliance(single/central IRB) page.
- Letters of support from entities providing resources or services for the study.
Other Requirements
- Requests to review medical records or charts will require HIPAA authorization to review Protected Health Information (PHI). PI's working with this type of information should also complete the compliance training modules conducted by the Compliance Office in the Howard University Hospital.
All researchers who wish to access data from the Rosemary Williams Cancer Registry (HU Cancer Center Registry) must first complete the Pre-Screening Interview Form in conjunction with the Manager of the Register. The approved data request form is submitted to the IRB along with the appropriate level data application. For more information on the cancer center registry, please visit the Rosemary Williams Cancer Registry website.
Defined waivers or alterations to informed consent or the documentation of informed consent, if applicable. The informed consent process may be waived or altered under certain circumstances outlined in 45 CFR 46.116(d). The documentation of informed consent may be waived under the circumstances outlined in 45 CFR 46.117(c). A workflow is available to determine if the waiver or alteration of informed consent or the documentation of informed consent are possible.
Just-In-Time (JIT) Notifications
JIT notifications sometimes come with a request to provide a rapid turn-around for IRB review. JIT notices must be read carefully so that the requirements are understood.
JITs do not warrant immediate review and will be scheduled for review at the next available IRB meeting. The investigator must update the funding entity of study's expected IRB review date.
Education and Resources
Education
Resources
Forms and Tools
- iRIS Platform
- AI Policy
- Ethical IRB Guidelines and Principles
- GCP Training Requirement
- Guidance for Case Reports
- Guidance for Faculty and Research Staff
- Guidance on Recruitment Material
- Guidance: Student Research Guide
- IRB Charge Policy
- IRB Communication with the RSC
- IRB SOP
- IRB SOP Supplement
- IRB SOP DoD Addendum
- Limited IRB Review
- Review of PHI-Related Protocols
- Template: Assent Form (Download)
- Template: Informed Consent Form (Download)
- Template: Interview Preamble (Download)
- Template: Survey Preamble (Download)
- Typical IRB Review Timelines
{kind=link}
Links
- 45 CFR 46: The Common Rule | eCFR
- 21 CFR 50: FDA regulations on the protection of human subjects | eCFR
- 21 CFR 56: FDA regulations on Institutional Review Boards (IRBs) | eCFR
- A How to Guide: The Use of Social Media in Research | NIH
- Belmont Report
- Human Research Protection Program Resources | HHS
- Office for Human Research Protections (OHRP) | HHS